Genomic medicine centers around a deceptively simple question: how do genetic variations affect our health? This is the problem of variant effect prediction, where "effects" can mean anything from impacts on protein function to implications for disease. Understanding variant effects lets us diagnose genetic conditions, offer more accurate prognoses, and decide on the best treatment for each individual.
But variant effect prediction is a difficult task because the human genome is complex and only partially understood. There are millions of known variants scattered across billions of base pairs, many of which are unique to individual patients.
As a result, genetic diagnosis is often inconclusive. Consider a child with symptoms of a rare genetic condition: after genetic sequencing, a genetic counselor cross-references the child's variants against ClinVar, a database of millions of variants and their effects. But most entries are labeled only as variants of uncertain significance (VUS), i.e., their effect on health is unknown. [3]ClinVar Miner: Demonstrating utility of a Web-based tool for viewing and filtering ClinVar data
Henrie A, Hemphill SE, Ruiz-Schultz N, et al. Hum Mutat. 2018 Aug;39(8):1051–1060. [DOI] A VUS classification gives clinicians little to act on, and can put patients in diagnostic limbo for years.
Computational approaches for variant effect prediction help, but most tools are limited to coding regions or can't explain their predictions – so don't always translate to clinical utility. However, the broader landscape is shifting. Genomic medicine is expanding rapidly as sequencing costs fall and clinical adoption grows. At the same time, a new generation of genomic foundation models – trained on vast numbers of DNA sequences – are capturing deep biological structure in their internal representations. And as these models grow more capable, interpretability methods are becoming increasingly useful, giving us tools necessary to understand the information that the models contain.
In this post, we describe research (which is undergoing peer review) done as part of our ongoing collaboration with Mayo Clinic. We show that embeddings from Evo 2, a genomic foundation model, support a new approach to variant effect prediction which is accurate across all variant types, generalizable to held-out data, and able to generate mechanistic hypotheses at scale.
We tackle two related problems:
- Pathogenicity prediction, i.e., which variants are disease-causing?
- Variant effect prediction, i.e., which biological functions are disrupted?
Our pathogenicity probe achieves state-of-the-art performance (0.997 overall AUROC on 839k ClinVar variants), outperforming existing approaches. Taken together, we think our results suggest a meaningful shift in how variant effect prediction can be done at scale – using interpretability tools as the translation layer between genomic foundation model embeddings and the vocabulary of currently known biological concepts. As a first step towards that, we're releasing the variant effect predictions and hypothesized explanations for all 4.2 million ClinVar variants through the Evo Variant Effect Explorer (EVEE), an interactive web resource for the broader research community.
Computational predictions are not diagnoses. But better computational tools mean better inputs to clinical decisions – and a more interpretable, genome-wide approach to variant effect prediction is a significant advance in what these tools can do.
Contents
Background: Evo 2, a foundation model for understanding the genome
Evo 2 is a large neural network trained on billions of DNA sequences spanning all domains of life. Like large language models trained on text, it develops rich internal representations of its training data – in this case, learning to encode the functional logic of genomes: e.g., which sequences are conserved, where genes begin and end, and how regulatory signals are structured. These representations span both coding and non-coding regions, meaning Evo 2 isn't subject to the coverage limitations that constrain RNA or protein-based models.
These rich representations can be leveraged even for tasks Evo 2 was not explicitly trained on. In fact, Evo 2 had not seen any human genetic variation at train time, meaning the results described here rely entirely on the deep evolutionary priors it learned from genomes across all domains of life. Rather than training a new model from scratch on limited variant annotation data, we can use Evo 2's embeddings as a foundation – a compressed encoding of everything the model has learned about genomic function – and build lightweight classifiers on top of them. For example, this approach is quite successful at predicting exon labels [1]Genome modelling and design across all domains of life with Evo 2
Brixi, G., Durrant, M.G., Ku, J. et al. Nature (2026). [DOI]. This approach generalizes well because the hard work of understanding the genome is already encoded in the representations. And because we're working directly with those representations, we can use interpretability tools to explain our predictions based on the biological features disrupted.
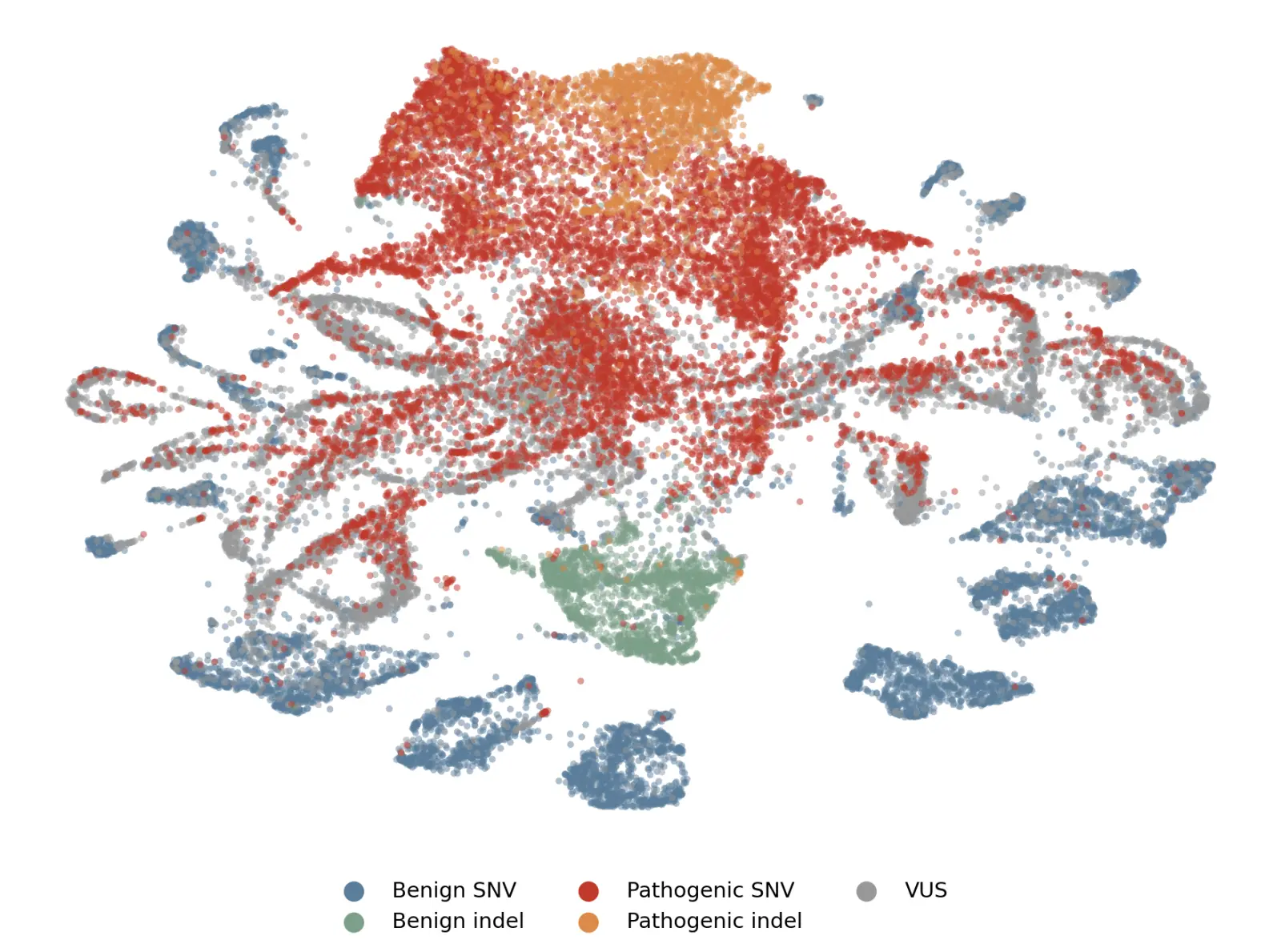
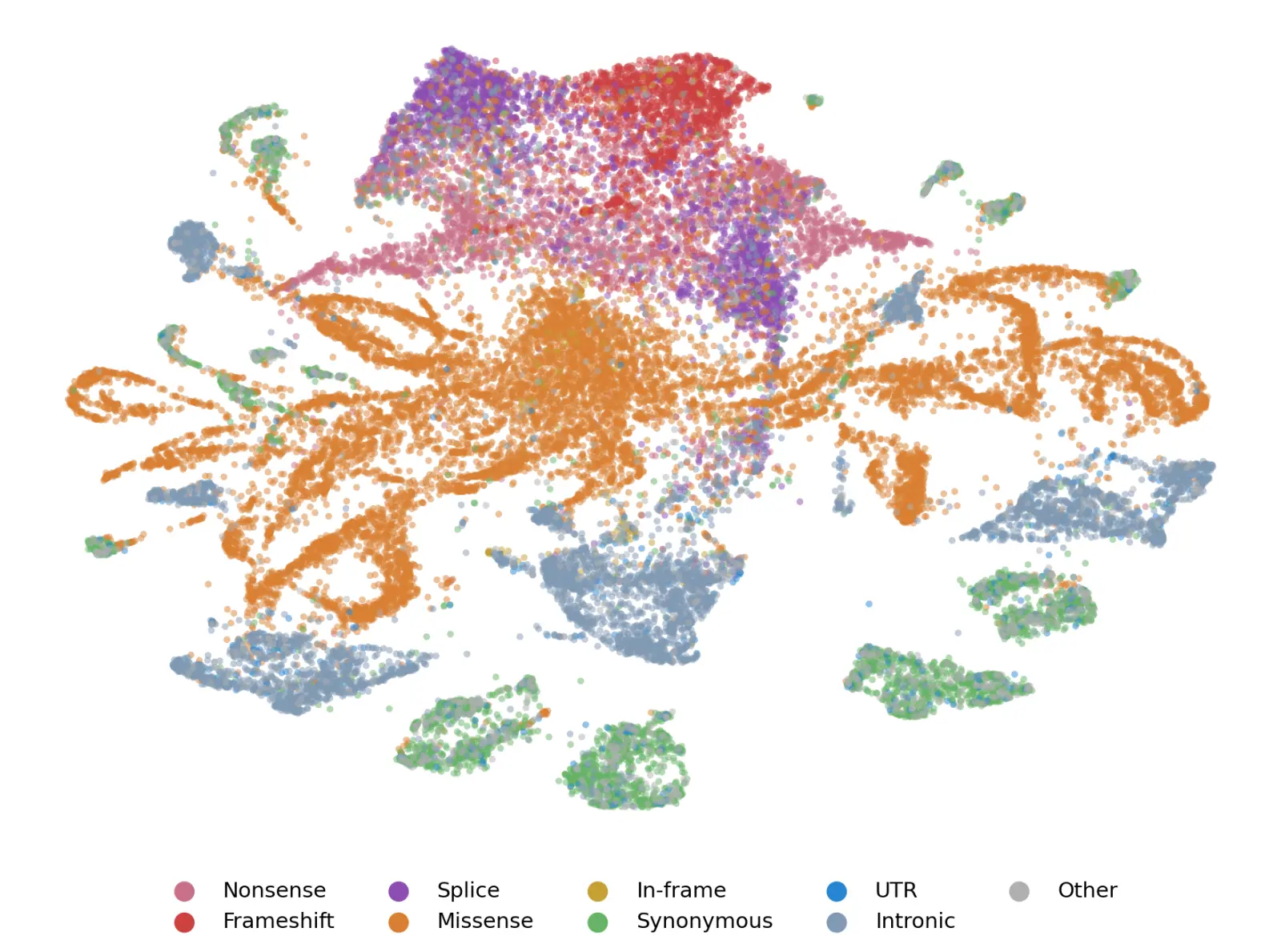
Probing for pathogenicity
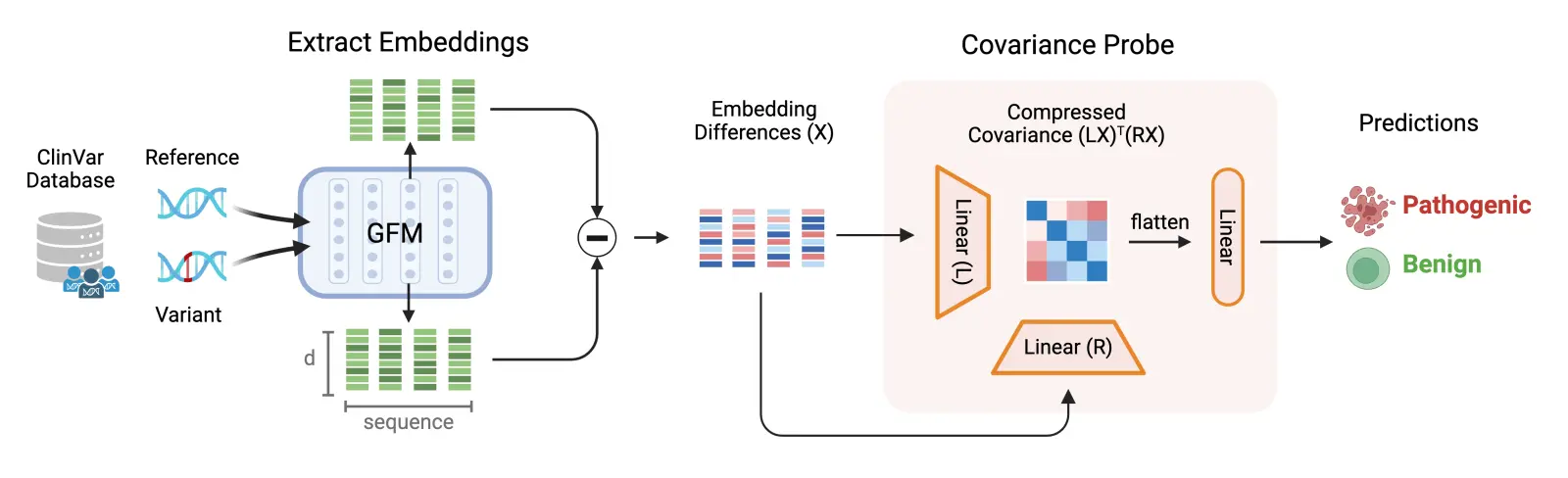
Our first contribution is a single, genome-wide pathogenicity probe: a lightweight classifier trained on top of frozen Evo 2 embeddings to predict whether a variant is likely to cause disease. Rather than training a new model end-to-end on variant annotation data, which is limited in scale and coverage, we treat Evo 2's representations as a foundation and learn to read pathogenicity directly off of them.
A key architectural choice is how we pool information across a sequence. Standard approaches use mean pooling, which blurs features across positions, discarding relational structure. We instead use covariance-based sequence pooling [2]Covariance-based Sequence Pooling
Dooms, T., Wang, N. K., and Pearce, M. T. Goodfire Research, 2026. [link], which captures feature co-occurrence (e.g., whether a particular motif appears within a specific structural context).
The probe achieves 0.997 AUROC on 839k ClinVar single nucleotide variants and generalizes zero-shot to short insertions and deletions (indels) with 0.991 AUROC – variant types it was not explicitly trained on. It outperforms classical meta-predictors, protein-based models, and existing foundation model approaches across consequence types and conservation levels, and transfers to deep mutational scanning datasets for BRCA1, BRCA2, TP53, and LDLR.
To understand why this matters, it helps to understand where existing tools fall short. Protein-based methods like AlphaMissense are restricted to missense variants and can't see the large fraction of disease-causing mutations that act through regulatory or splicing mechanisms. CADD is genome-wide but aggregates over 100 annotation features with crossed terms and rank transformations that make individual contributions hard to disentangle. Our approach sidesteps both limitations: because it operates on DNA directly, it covers the full spectrum of variant types; and because we also probe for relevant biological features directly, we can predict how each is disrupted.
Explaining 4.2 million variants
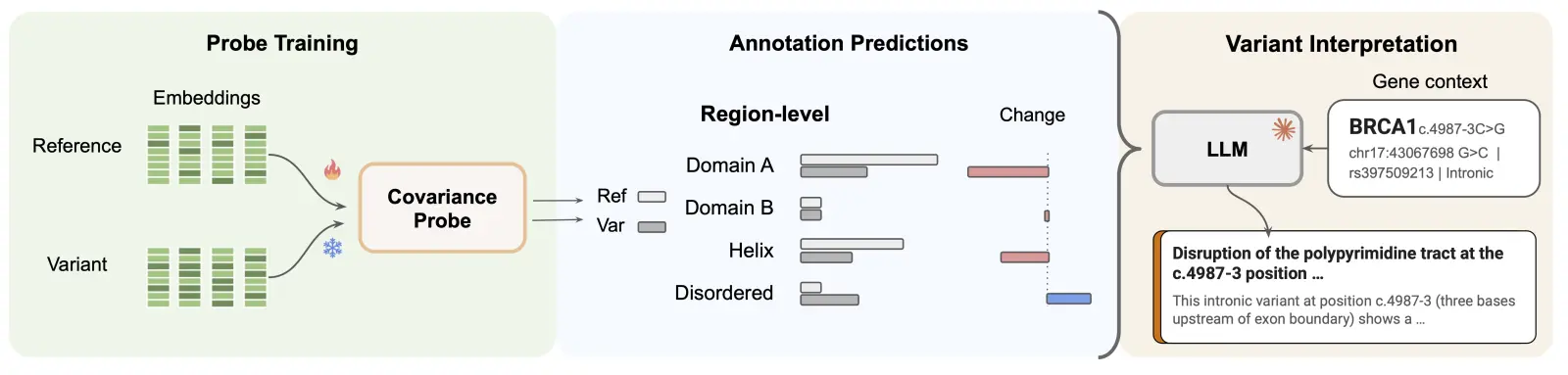
A pathogenicity score is useful, but the more valuable question to researchers and clinicians is how a variant disrupts downstream biology, e.g. which functional elements it affects, or which molecular mechanisms are implicated. This is the harder problem of variant effect prediction proper, and it's where interpretability becomes central.
Our pipeline works in two stages. First, we train probes on reference sequence embeddings – each probe predicting known biological annotations such as splice sites, regulatory elements, and structural domains – and then quantify how much each variant shifts those predictions relative to the reference. This produces a disruption profile, a structured summary of which biological functions a variant is likely to perturb. Second, we use frontier reasoning models to synthesize those disruption profiles, combined with the pathogenicity prediction and context on the surrounding gene, into natural-language explanations, grounding each explanation in the specific evidence extracted from Evo 2's representations.
The framing we find useful here is one of translation. Evo 2's embeddings are information-dense but not directly legible: they encode a great deal about genomic function, but in a form that doesn't map onto the vocabulary that we human scientists use to reason about biology. The annotation probes act as a translation layer, converting those representations into structured predictions over known biological concepts like splice disruption or domain interference. The language model then synthesizes those predictions into something a researcher can read and evaluate.
To quantify explanation quality, we ran an LLM-as-a-judge evaluation against 154 high-confidence ClinVar variants (≥3 review stars), scoring each synthesized interpretation on mechanism coverage, biological accuracy, and specificity. Our approach leverages Evo 2's predicted disruption profiles to achieve a composite score of 3.8/5, improving significantly over the 2.8/5 from variant metadata alone.
The practical output of this work is the Evo Variant Effect Explorer (EVEE): an interactive resource providing pre-computed pathogenicity scores, disruption profiles, and on-demand natural-language explanations for all 4.2 million variants in ClinVar – including roughly 2 million currently classified as variants of uncertain significance. For the first time, every variant in the database has a structured, human-readable hypothesis about its potential mechanism. These are computational predictions, not clinical conclusions, but we hope EVEE serves as a resource that can help inform researchers and clinicians, and a foundation for future work that pushes interpretable variant effect prediction further.
EVEE is available to the community as a freely accessible web resource [link].
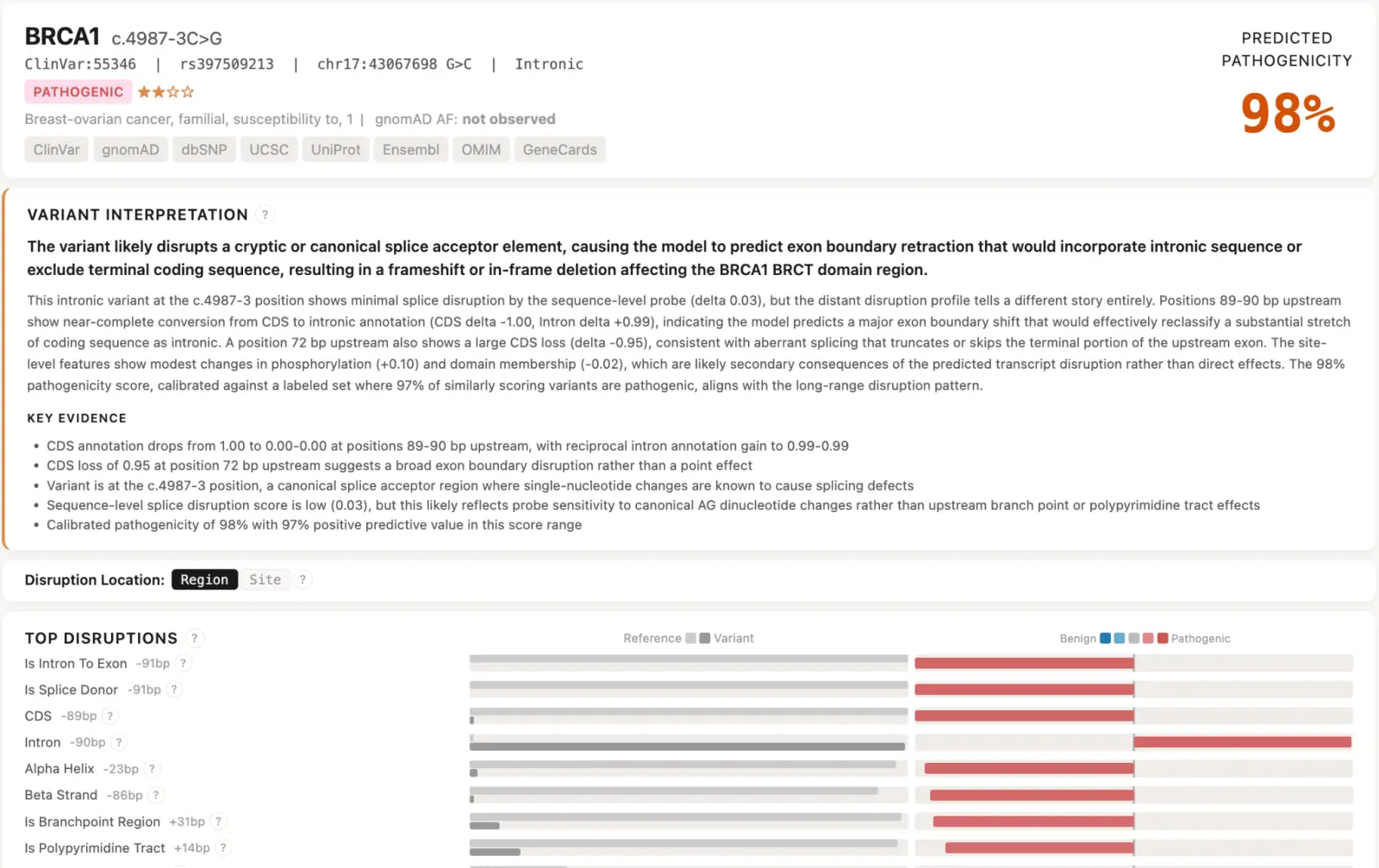
What this points toward
Leveraging frontier models in both biology and natural-language reasoning, we present an approach to variant effect prediction that achieves state-of-the-art performance, genome-wide coverage, and interpretable-by-design predictions for all 4.2 million variants in ClinVar.
Most computational approaches to variant effect prediction work downstream: they reason about proteins, regulatory scores, or other derived signals. Working directly at the DNA level, as we do here, means operating closer to the mutation itself. The conventional tradeoff is that upstream approaches tend to generalize better but perform worse on any given task. The results here suggest that with a sufficiently capable genomic foundation model, that tradeoff can be broken: genome-wide coverage and strong performance are not mutually exclusive.
A few limitations are worth flagging:
- The predictions described here are still hypotheses, and experimental validation is the necessary next step.
- The annotation probes can only surface known biological mechanisms – truly novel ones would fall outside their scope; unsupervised techniques like sparse autoencoders could complement our approach and discover as-yet-unnamed functional features.
- The natural-language explanations generated by the reasoning models should be treated as hypotheses requiring expert review, not as clinical conclusions.
- The research described here is a preprint and is currently undergoing peer review.
At the same time, the approach developed here could establish a methodological foundation for tackling the more complex, polygenic effects that underlie common disease.
More broadly, we believe this points toward a general paradigm. A central challenge in genomics is turning rich but opaque representations into structured, human-interpretable knowledge. This challenge is not unique to the field, and interpretability tools address it directly: they are a translation layer between the internal representations of neural networks and a vocabulary of structured, human-readable concepts. Wherever powerful foundation models encode rich scientific knowledge, we believe this approach offers a principled path from embeddings to scientific understanding.