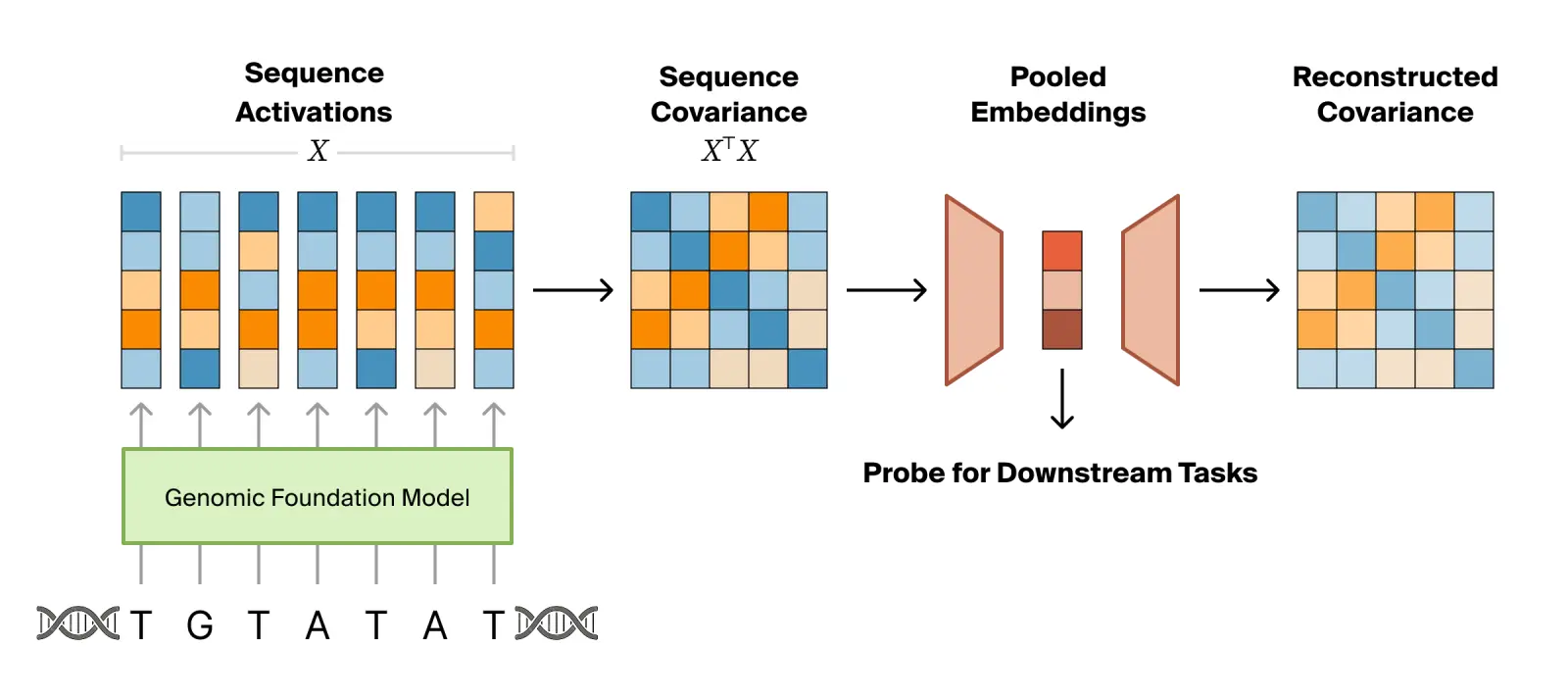
Genomic foundation models like Evo 2 [1]Genome modelling and design across all domains of life with Evo 2
Brixi, G., Durrant, M.G., Ku, J. et al. Nature (2026). https://doi.org/10.1038/s41586-026-10176-5, AlphaGenome [2]Advancing regulatory variant effect prediction with AlphaGenome
Avsec, Ž., Latysheva, N., Cheng, J. et al. Nature 649, 1206–1218 (2026). https://doi.org/10.1038/s41586-025-10014-0, and NTv3 [3]A foundational model for joint sequence-function multi-species modeling at scale for long-range genomic prediction
Sam Boshar, Benjamin Evans, Ziqi Tang et al. bioRxiv 2025.12.22.695963. https://doi.org/10.64898/2025.12.22.695963 learn sequence embeddings rich enough to predict protein properties, gene expression levels, and mutational effects from DNA alone. Yet the biological unit that matters for most applications is the gene, not the individual nucleotides, so per-token embeddings must be aggregated over thousands of positions. Mean pooling is the field default: simple, stable, and surprisingly competitive. But it is structurally blind to how features relate to each other within a sequence, which matters whenever combinations of features are important.
We propose covariance pooling, which uses the truncated second moment of token activations rather than the first, as a principled replacement that can be used for both supervised probing and learning unsupervised embeddings. The second moment preserves the joint activation structure that the mean discards, captures the shape of the per-token cloud of embeddings rather than just its centroid, and requires no labels or architectural choices. By training to reconstruct the second moment, we can learn a low-rank factorization that compresses the sequence of activations into a compact embedding.
Across Gene Ontology prediction and genomic track prediction on NTv3, covariance pooling strongly outperforms mean pooling. We argue it should be the new baseline for gene-level probing of genomic foundation models as well as other sequence-based modalities.
Contents
Background
Pooling mitigates overfitting when training on foundation model activations.
Genomic foundation models are trained on massive corpora of DNA to learn the latent grammar of the genome in a self-supervised manner. Much like language models, they compress vast amounts of sequence data into rich internal representations.
However, effectively utilizing these representations for specific downstream tasks, particularly at the gene level, presents a unique set of obstacles. These foundation models natively output a sequence of activations, but biological function usually emerges at the scale of an entire gene. Translating a model's raw, token-level representations into an actionable prediction requires aggregating this information, a process hindered by several challenges:
- Data size: Gene lengths span multiple orders of magnitude from hundreds to millions of nucleotides. A single gene can consist of up to gigabytes of activations, meaning the gradient signal is weak and diffuse.
- Generalization: Probes must generalize to highly differing distributions so that results hold broadly, rather than overfitting to specific population sets (e.g., ancestry groups).
- Limited data: The downstream tasks we are interested in often have very limited data, either because labels are expensive to acquire or because the dataset is inherently capped (e.g., the human genome only has about 20,000 genes).
This combination of massive inputs and sparse labels requires being incredibly careful about overfitting. The default practice in the field is to apply mean pooling over the sequence activations to create a constant-size embedding on which a linear probe is trained. This method acts as a strong regularizer and often achieves strong baseline generalization on downstream tasks [4]Benchmarking DNA foundation models for genomic and genetic tasks
Feng, H., Wu, L., Zhao, B. et al. Nat Commun 16, 10780 (2025). https://doi.org/10.1038/s41467-025-65823-8, but leaves critical structural information behind.
Mean pooling discards the combinatorial logic of genomic regulation.
Genomic foundation models process sequences into per-token embedding vectors $X \in \mathbb{R}^{T \times d}$. While simply aggregating the presence of features via mean pooling is a safe baseline, many downstream tasks can rely on combinations of features at the same location that mean pooling fails to capture.
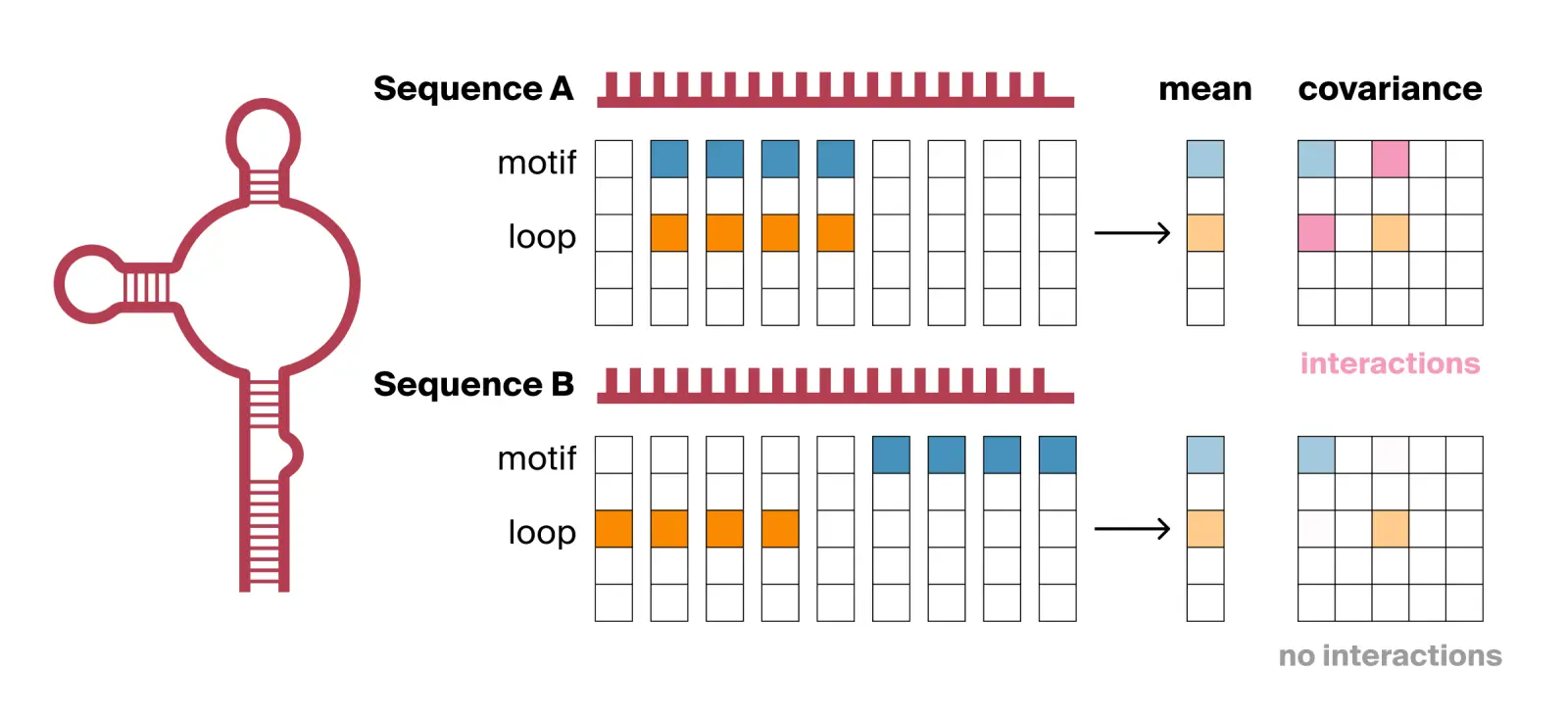
For example, many RNA-binding proteins only bind to an RNA transcript if a specific genetic sequence motif sits exactly within a specific physical shape, such as an accessible, unpaired loop. Imagine a foundation model encoding such a sequence, where one feature dimension detects the target sequence motif and another detects the hairpin loop structure. Consider two RNA sequences that both contain the motif and loop structure (see Fig. 2):
- Sequence A (bindable): The sequence motif occurs exactly at the loop. The model encodes this physical overlap into a single sequence token where the motif and loop feature dimensions fire strongly at the same time.
- Sequence B (not bindable): The sequence motif is buried in a tightly paired structural stem, while an unpaired loop made of the wrong sequence sits elsewhere. The model encodes these separated features in entirely different sequence tokens.
If we apply mean pooling, $$\mu = \frac{1}{T} X^\top \mathbf{1}_T,$$ the model averages these dimensions independently across the entire sequence. It simply tallies the isolated properties and yields the exact same centroid for both sequences, destroying the information about the physical intersection required for biological function.
While all pooling strategies are ultimately bound by the foundation model's native encoding, mean-pooling inevitably erases combinatorial structure regardless of the representation. For instance, even if the model explicitly encodes a specific “feature disrupted” signal for Sequence B, averaging detaches that status from its spatial context, leaving it unclear which motif or loop was inactivated. Ultimately, mean pooling prematurely destroys whatever structural logic the foundation model successfully preserved.
Covariance pooling
Motivation: Covariance pooling better preserves sequence structure
The second moment explicitly preserves this structural logic: $$C = \frac{1}{T} X^\top X.$$ Its $(i,j)$ entry measures how often feature dimensions $i$ and $j$ are large at the same time within the same token. Because it computes the token-by-token outer product, its off-diagonal entries natively surface whether two dimensions co-activate. If the sequence and loop occur together (Sequence A), their product is highly positive. If they occur separately (Sequence B), their product at any given position is effectively zero. Due to linearity, the second moment can capture this structure for any pair of linearly accessible feature directions, not just the model's residual stream dimensions.
We can see why this works mathematically by writing the (compact) singular value decomposition (SVD) of our activation matrix as $$X = U \Sigma V^\top.$$ When we compute the second moment, it factors to: $$C = \frac{1}{T} V \Sigma^2 V^\top.$$ The matrix $U$, which dictates which specific token maps to which direction, cancels out due to its orthogonality. What survives is $V$ (which directions the tokens span) and $\Sigma$ (the variance magnitude per direction). The covariance captures the shape of the token cloud while discarding token identity and absolute position—i.e., it encodes the relational information about the sequence without actually encoding the sequence itself.
Learning embeddings with and without supervision
While the raw second moment captures useful structural relationships, using it directly is impractical in terms of storage and prediction. Modern foundation models have massive hidden dimensions, meaning a full covariance matrix would contain millions of entries. Since many features are redundant or capture trivial coincidences, we filter this noise by projecting the raw token activations through a learnable bottleneck. This forces the embedding to retain important features, yielding a compact matrix that represents only the most dominant regulatory interactions: $$M = \frac{1}{T} \left(XL\right)^\top \left(XR\right)$$
Introducing these projection layers transforms the pooling step from a static mathematical operation into a learnable operation. We can now teach the model which features are important enough to keep in our condensed matrix. If we have specific functional labels, we can train this bottleneck and downstream probe end-to-end in a supervised manner. As the downstream probe learns, gradients flow directly back into the projection weights. This allows the bottleneck to prioritize the specific structural overlaps that drive that label and ignore the irrelevant background data.
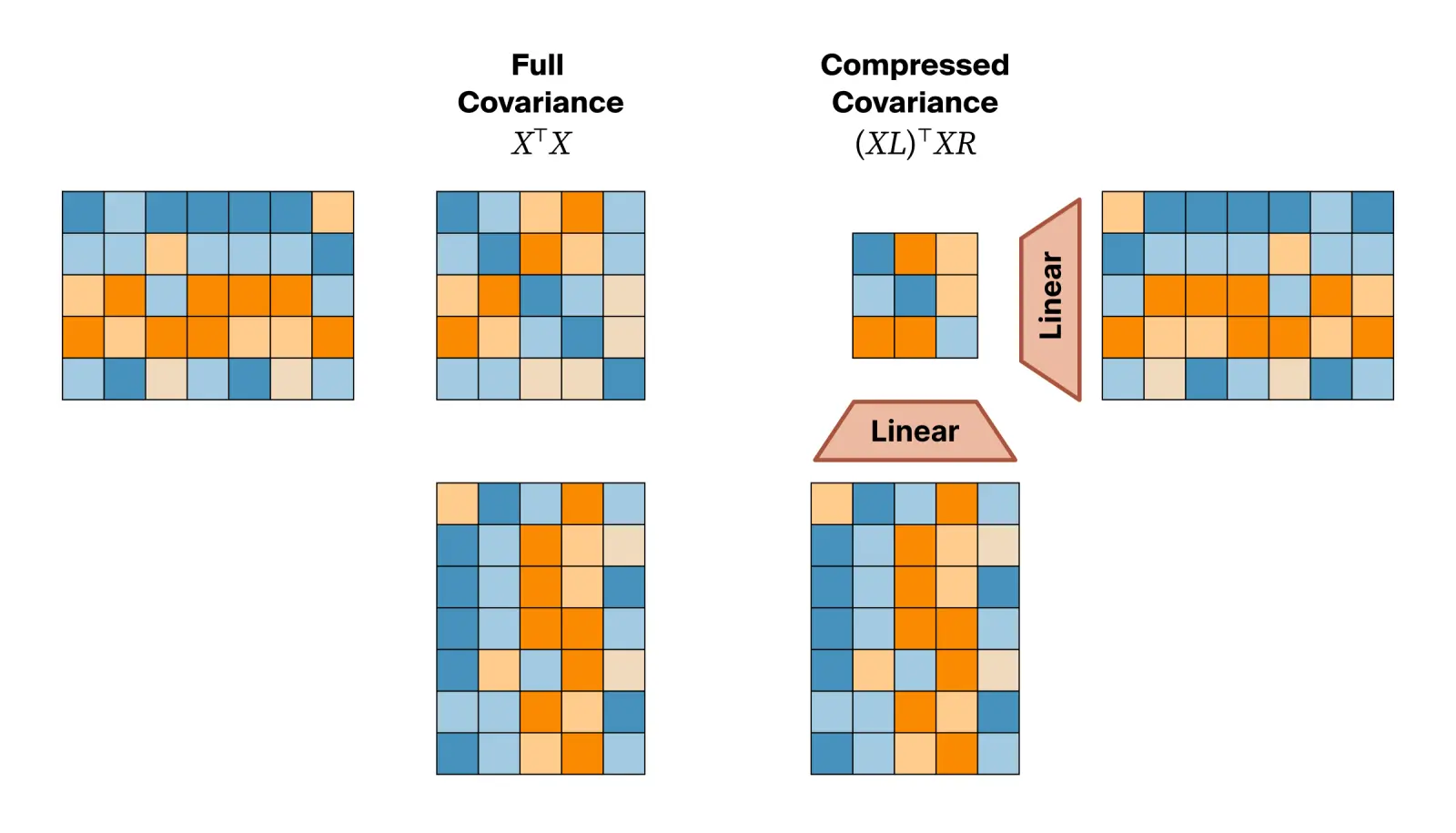
Alternatively, these embeddings can be trained entirely without labels to create a task-agnostic embedding. In this unsupervised setting, we treat the linear projections as a sequence-based autoencoder. Standard token-level autoencoders minimize the token-level reconstruction error, which effectively maximizes the trace of a token-to-token similarity matrix so that each token predicts itself. Instead, we task the bottleneck with reconstructing the sequence's full covariance. This objective captures the joint activation structures that pointwise reconstruction misses.
While this would naively require constructing the sequence-by-sequence matrix, which is computationally infeasible for long genes, we can use a simple mathematical equivalence to avoid this. Maximizing the Frobenius norm of the sequence-level reconstruction is practically similar to maximizing the norm of the d_model-by-d_model covariance matrix:
$$\|X^\top X\|_F = \|XX^\top\|_F.$$
This means we can efficiently optimize sequence-wide reconstruction in linear time. In short, we get the benefits of a sequence autoencoder without complications like positional embeddings and explicitly defined decoders.
Despite the name “covariance pooling”, “second-order pooling” would arguably be more precise, as $\hat{C}$ is not necessarily symmetric (since $L$ and $R$ are untied), and we didn't find subtracting the mean or any related augmentation to yield improvements.
Implementation
Both the supervised and unsupervised version of covariance pooling can be implemented in a few lines of PyTorch code.
# X: [seq_len, d_model]
# L, R: [d_model, d_compressed]
# seq_len: sequence length
# d_model: hidden dimension of model
# d_compressed: probe hidden dimension
# probe on the embedding or a flattened version
emb = (X @ L).mT @ (X @ R) / seq_len # [d_compressed, d_compressed]
sup_loss = probe(emb.flatten())
# reconstruct through the linear bottleneck
target = (X.mT @ X) / seq_len # [d_model, d_model]
recons = L @ emb @ R.mT # [d_model, d_model]
unsup_loss = torch.norm(recons - target, p="fro")Results: Covariance pooling outperforms mean pooling on downstream tasks
To validate whether this learnable bottleneck extracts more functionally relevant information than mean pooling, we evaluated the embeddings across two distinct genomic tasks using the NTv3 model:
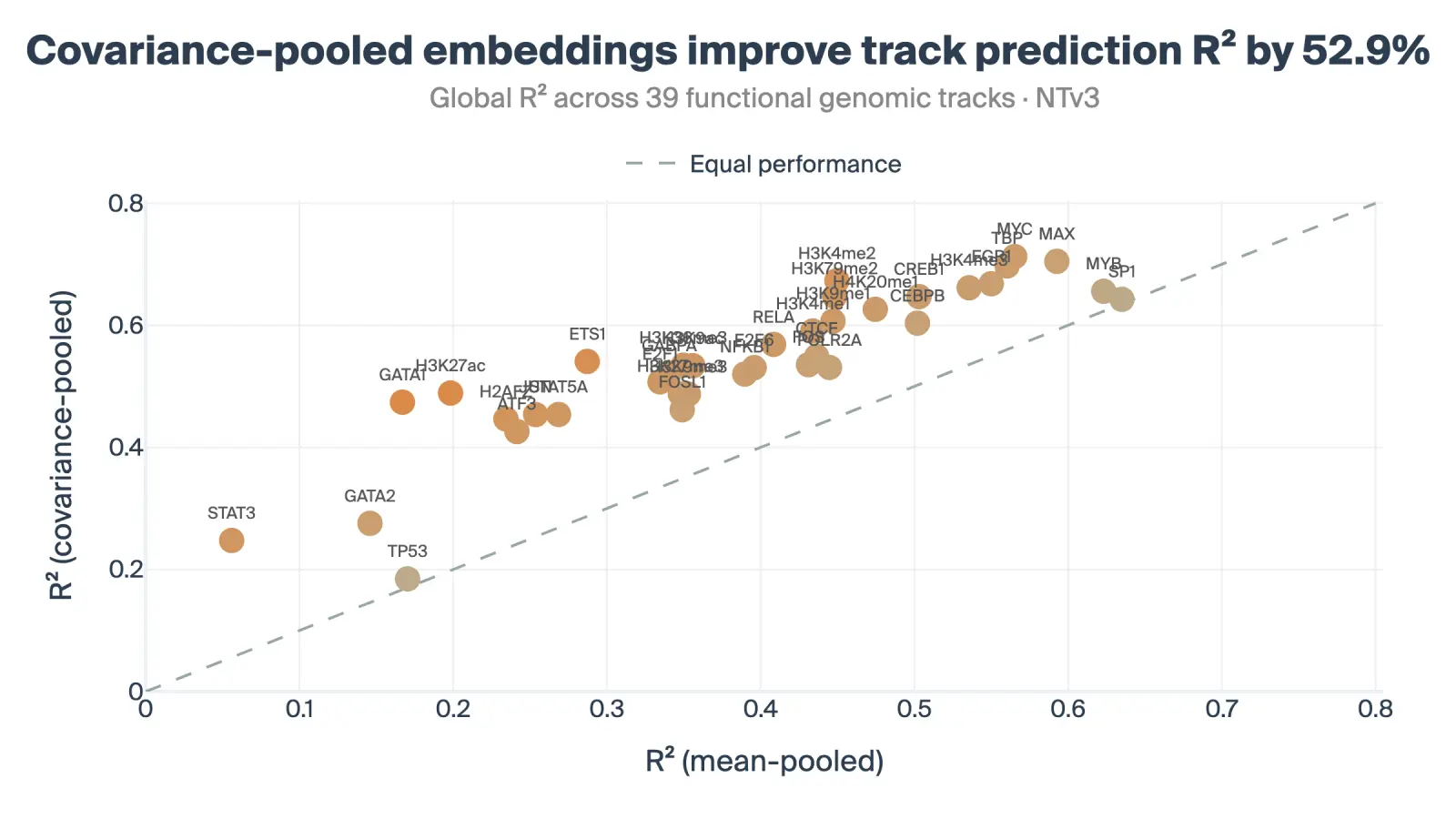
Genomic Track Prediction: We trained probes to predict the mean signal of 39 diverse functional genomic tracks. This task measures whether the embedding successfully encodes dense regulatory information like chromatin accessibility and transcription factor binding. The learned covariance-based embeddings improved the track prediction R² by 52.9% over the mean-pooling baseline.
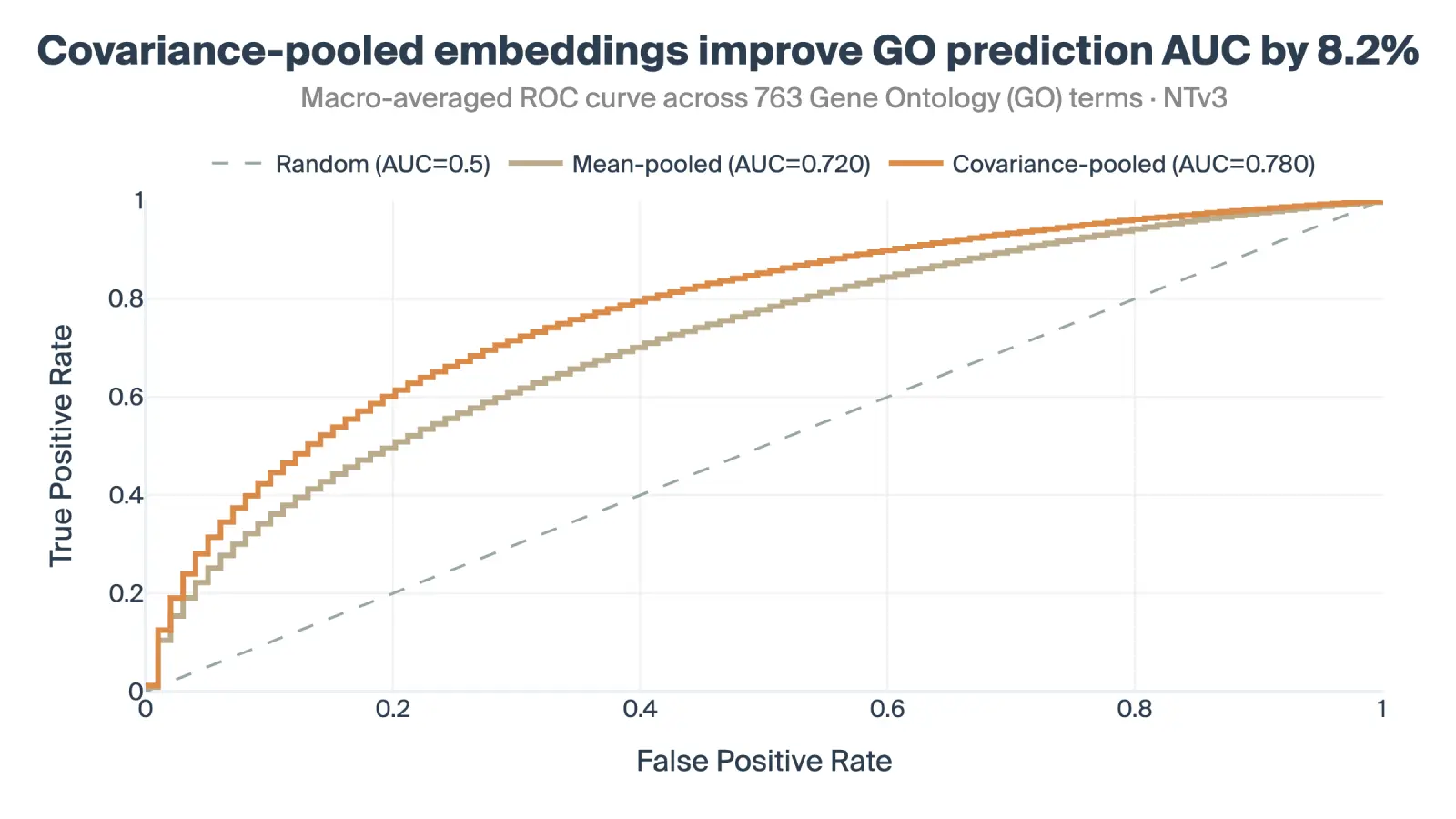
Gene Ontology (GO): We filtered the Gene Ontology database for a diverse set of 763 biological terms. Predicting these functional labels using the frozen, unsupervised autoencoder embeddings yielded an 8.4% AUC lift over the mean-pooled baseline.
The above are small-scale experiments to sanity-check the approach. Nevertheless, forthcoming work will show that this approach scales gracefully to larger datasets.
Covariance pooling as a baseline for sequence aggregation
Mean pooling became the default because it's simple, stable, and acts as a strong regularizer for huge activation sequences. As genomic foundation models become increasingly capable, they learn intricate representations that mean pooling discards, leaving structural blind spots to important positional mechanisms in the sequence.
Covariance pooling offers a principled, drop-in replacement that is more informative. When paired with a learned bottleneck and a global reconstruction objective, it compresses gigabytes of raw sequence activations into a compact, stable embedding, sidestepping the overfitting traps of more complicated attention-based poolers in low-data regimes.
Existing bioinformatic pipelines already set a high bar for genomic annotation prediction; covariance pooling will not replace them overnight, but promises to improve how we extract knowledge from foundation models to match that bar. As genomic models improve, extracting the native richness of their representations will become increasingly important to fully unlock their biological utility. Covariance pooling is a strong baseline that achieves this without massive datasets or architectural overhauls.
We believe unsupervised discovery through foundation models will play a huge role in advancing scientific knowledge, and our tooling needs to be designed to express that potential. These interpretability-based approaches turn foundation models into engines of scientific discovery, enabling us to understand how these models think about complex domains, which we will be exploring in forthcoming work.